Chapter 11 Emner fra eksperimental design
library(tidyverse)
library(broom)“Amatører sidder og venter på inspiration, resten af os står bare op og går på arbejde.” - Stephen King
11.1 Inledning og læringsmålene
11.1.1 Læringsmålene
I skal være i stand til at
- Beskrive randomimisation, replication and blocking
- Beskrive Simpson’s paradox
- Beskrive Anscombes quartet
- Tjekke efter batch effects med PCA
11.1.2 Inledning til chapter
Formålet med dette kapitel er at spørge: hvordan kan vi anvende værktøjerne som vi har lært i kurset til at kigge nærmere på forskellige emner i eksperimental design? Det er slet ikke en grundig introduktion til eksperimental design, men nogle nyttige og også interesseret emner som godt viser hvorfor det er vigtigt at lave hensigtsmæssige visualiseringer af dit data. Forståelsen af hvordan batch effekts påvirker en analyse er særlig vigtig indenfor biologi-fag, hvor mange store sekvensering projektor involverer data samlede eller sekvenseret over forskellige batches, sekvensering maskiner eller forskellige forberedelsesmetoder.
11.1.3 Video ressourcer
- Part 1: randomisation, replikation, blocking + confounding
- Ingen video: læs gerne notaterne nedenfor
- Part 2: Simpson’s paradox
Link her hvis det ikke virker nedenunder: https://player.vimeo.com/video/556581563
- Part 3: Anscombe’s quartet
Link her hvis det ikke virker nedenunder: https://player.vimeo.com/video/556581540
- Part 4: Batch effects and principal component analysis.
OBS Man kan selvfølgelige også anvende map_if() til at log-transformere.
Link her hvis det ikke virker nedenunder: https://player.vimeo.com/video/556581521
11.2 Grundlæggende principper i eksperimental design
11.2.1 Randomisation and replication
Man laver et eksperiment for at få svar på et bestemt spørgsmål, eller hypotese. Og man designer eksperimentet ud fra principper som gøre det gyldigt at fortolke resultaterne fra analysen af datasættet bagefter. For et eksperiment at være gyldigt skal det kunne demonstrere hensigtsmæssige replikation og randomisation.
Randomisation
Man vil gerne udelukke, at konklusionerne kan bare skyldes variansen pga. en faktor som ikke direkte er interessant i eksperimentet. Derfor bruger man randomisation til at få disse faktorer fordelte over de forskellige treatment grupper. Et eksempel kan være ‘double-blinding’ i kliniske eksperimenter - både lægen og patienten har ikke kenskab til, hvem der hører til de forskellige grupper, og så kan man undgår forskelsbehandling indenfor grupperne, som kan påvirker de endelige resultater.
Replikation
Når man gentager et eksperiment flere gange - for eksempel ved at have flere patienter i hver treatment gruppe. Det tillader os at kunne beregne variabiliteten i data, som er nødvendig for at konkludere om der er en forskel mellem de grupper. Man kan altså ikke generalisere resultater som er blevet målt på kun én person.

Figure 11.1: randomisation og replikation
I ovenstående figur er der 6 replikater i hver gruppe, som er enten “treatment” eller “control”. I tilfældet “Good randomisation” er genstande som er taget tilfældige fra populationen vel matchet mellem de to grupper, mens i andet tilfældet “Poor randomisatin”, kan man se, at farverne af genstandene er vel matchet indenfor samme grupper. Det gøre det derfor umuligt at fortæl, om en eventuelle forskel mellem “treatment” og “control” er i virkeligheden resultatet af farven i stedet for målingerne, at man gerne vil sammenligne.
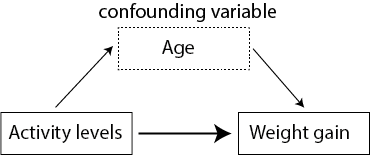
Confounding
Figuren nedenfor illustratorer age som counfounding variabel i et eksperiment hvor man prøver at forstå sammenhæng mellem aktivitet niveau og vægtøgning. Det kan være, at det ser umiddelbart ud til at være, at et lavt aktivitetsniveau (afhængig variabel) forklarer vægtøgning (unaghængig variabel), men man er nødt til at tage ændre variabler i betragtning for at sikre, at sammenhængen ikke skyldes noget andet. For eksempel, gruppen med det højt aktivitetsniveau kunne bestå af personer, der er yngre end personerne i gruppen med det lavt aktivitetsniveau, og deres alder kan påvirke deres vægtøgning (måske på grund af forskelligeheder i stress niveauer, kost osv.).
Blocking
Man kan prøve at kontrollere for ekstra variabler som vi ikke er interesseret i gennem “blocking”. Man laver “blocking” ved først at identificere grupper af individuelle som ligner hinanden så meget som muligt. Det kan være for eksempel at tre forskellige forsker var med til at lave et stort eksperiment med mange patienter og forskellige treatment grupper. Vi er interesseret i om der er forskellen mellem de treatment grupper, men ikke om der er en forskel mellem forskernes behandling af patienterne. Derfor vil vi gerne ‘block’ efter forsker - kontrollere for dem som en “batch” effect. Man kan også block efter fk. sex, for at sikre at forskellen i treatment grupper skyldes ikke forskelligheder mellem mænd og kvinder. Man laver “blocking” som del af en lineær model efter data er samlet, men det er nyttige at tænke over det fra starten.
11.2.2 Eksempel med datasættet ToothGrowth
Et god eksempel på et godt eksperimental design er datasættet ToothGrowth, som er baserende på marsvin - de fik forskellige kosttilskud og doser og så fik de målte længden af deres tænder.
data(ToothGrowth)
ToothGrowth <- ToothGrowth %>% as_tibble() %>% mutate(dose = as.factor(dose))
summary(ToothGrowth)## len supp dose
## Min. : 4.20 OJ:30 0.5:20
## 1st Qu.:13.07 VC:30 1 :20
## Median :19.25 2 :20
## Mean :18.81
## 3rd Qu.:25.27
## Max. :33.90Her kan man se, at for hver gruppe (efter supp og dose) er der 10 marsvin - vi har således replikation over de grupper, og hver supp (supplement) har hver af de tre mulige værdier for “dose”. Hvis vi for eksempel ikke var interesseret i supp men kun dose, så kan man ‘block’ efter supp for at afbøde forskelligheder i effekten af de to supplements i supp.
ToothGrowth %>% dplyr::count(supp,dose) %>%
ggplot(aes(x=factor(dose),y=n,fill=factor(dose))) +
geom_bar(stat="identity") +
ylab("Number of guinea pigs") +
xlab("Dosage") +
facet_grid(~supp) +
theme_bw()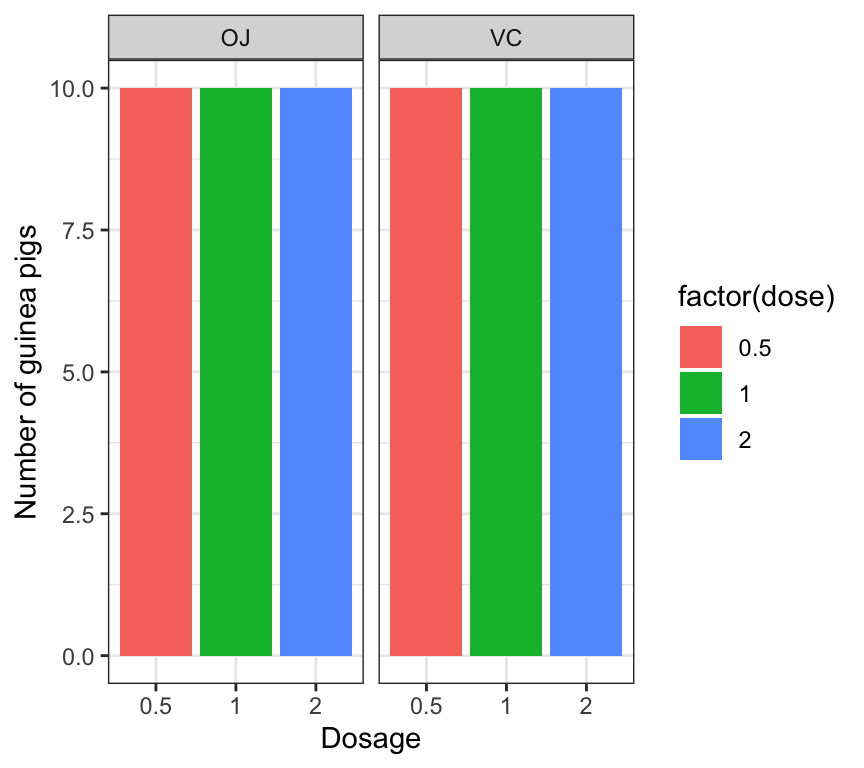
Man må dog passe på, fordi vi vide ikke om, hvordan de marsvin blev tilknyttet til de forskellige grupper. For eksempel, hvis male og female marsvin er ikke tilknyttet ved tilfælde, kan det opstå, at supp “OJ” og dose “0.5” har kun male guinea pigs og supp “OJ” med dose “1.0” har kun female guninea pigs. Så kunne vi ikke fortæl, om forskellen i dose “0.5” vs “1.0” er resultatet af de dose eller kønnet.
11.3 Case studies: Simpson’s paradox
(Se også videoressourcer Part 2).
Simpson’s paradox opstår når man drager to modsætte konklusioner fra det samme datasæt - på den ene side når man kigger på de data samlede, og på den anden side når man tager nogle grupper i betragtning. Vi kan visualisere Simpson’s paradox gennem eksemplet nedenfor - her har vi to variabler x og y som vi kan avende til at lave et scatter plot, samt nogle forskellige grupper indenfor variablen group.
#library(datasauRus)
simpsons_paradox <- read.table("https://www.dropbox.com/s/ysh3qpc7qv0ceut/simpsons_paradox_groups.txt?dl=1",header=T)
simpsons_paradox <- simpsons_paradox %>% as_tibble(simpsons_paradox)
simpsons_paradoxFALSE # A tibble: 222 × 3
FALSE x y group
FALSE <dbl> <dbl> <chr>
FALSE 1 62.2 70.6 D
FALSE 2 52.3 14.7 B
FALSE 3 56.4 46.4 C
FALSE 4 66.8 66.2 D
FALSE 5 66.5 89.2 E
FALSE 6 62.4 91.5 E
FALSE 7 38.9 6.76 A
FALSE 8 39.4 63.1 C
FALSE 9 60.9 92.6 E
FALSE 10 56.6 45.8 C
FALSE # … with 212 more rowsHvis vi bare ignorere group og ser på de data samlet, kan vi se at der er en stærk positiv sammenhæng mellem x og y. Men når vi opdele efter de forskellige grupper, ved at skrive colour = group, få vi faktisk en negativ sammenhæng indenfor hver af de grupper.
p1 <- simpsons_paradox %>%
ggplot(aes(x,y)) +
geom_point() +
geom_smooth(method="lm",se=FALSE) +
theme_classic()
p2 <- simpsons_paradox %>%
ggplot(aes(x,y,colour=group)) +
geom_point() +
geom_smooth(method="lm",aes(group=group),colour="black",se=FALSE) +
theme_classic()
library(gridExtra)
grid.arrange(p1,p2,ncol=2)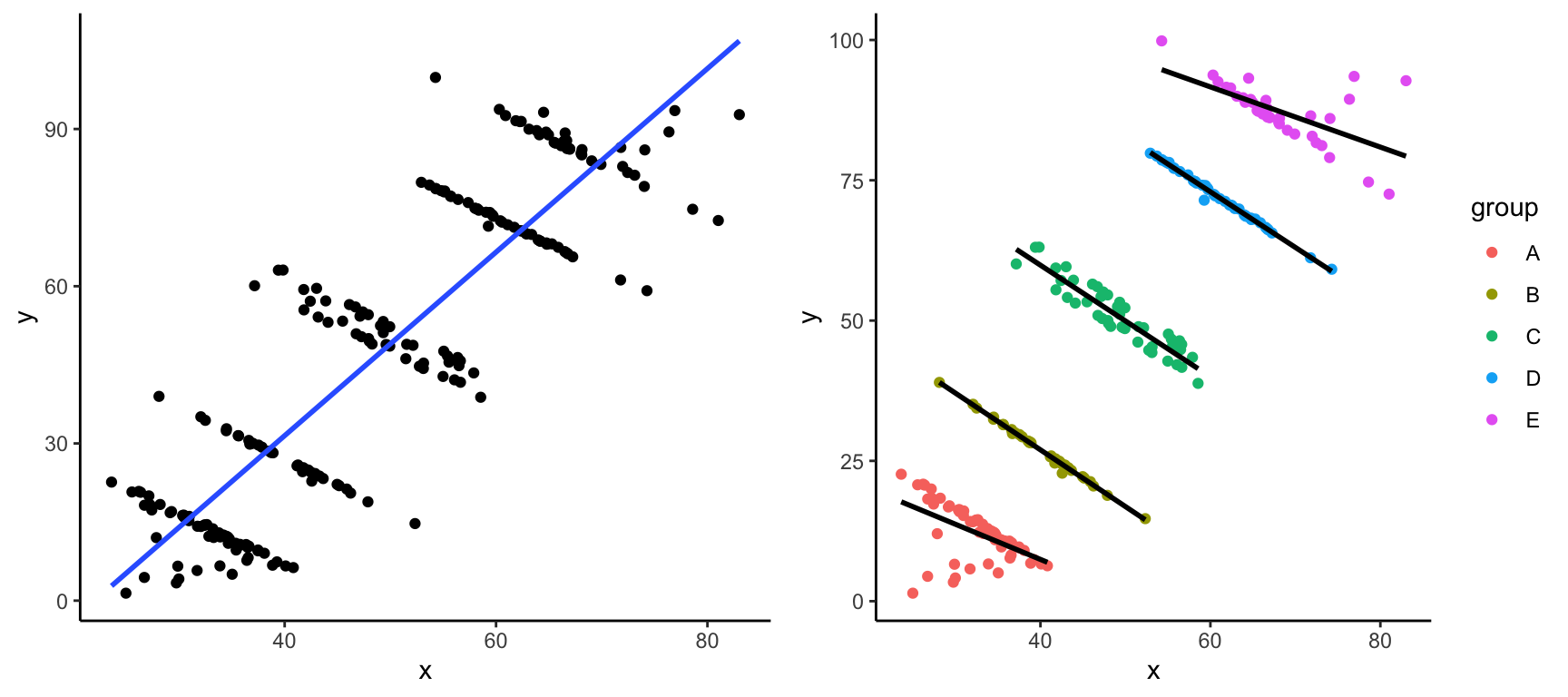
Simpson’s paradoks sker mere ofte end man skulle tror, og derfor er det vigtigt at tænke over hvad for nogen ændre variabler man også er nødt til at tage i betragtning.
11.3.1 Berkerly admissions
Det meste berømte eksempel af Simpson’s Paradox drejer sig om optagelsen til universitetet i Berkely i 1973. Følgende table fra Wikipedia (https://en.wikipedia.org/wiki/Simpson%27s_paradox) viser statistikker om antallet som ansøgt samt procenttallet som blev optaget overordnet i universitet efter kønnet.

Figure 11.2: source: wikipedia
Hvis vi lave et barplot af tallerne, kan man se, at der er en højere procenttal af mænd end kvinder som var blevet optaget på universitet (sagen medførte en retsag mod universitet).
admissions_all <- tibble("sex"=c("all","men","woman"),admitted=c("41","44","35"))
admissions_all %>% ggplot(aes(x=sex,y=admitted,fill=sex)) +
geom_bar(stat="identity") +
theme_minimal() +
ylab("Percent admitted") +
scale_x_discrete(limits = c("woman","men","all")) +
coord_flip()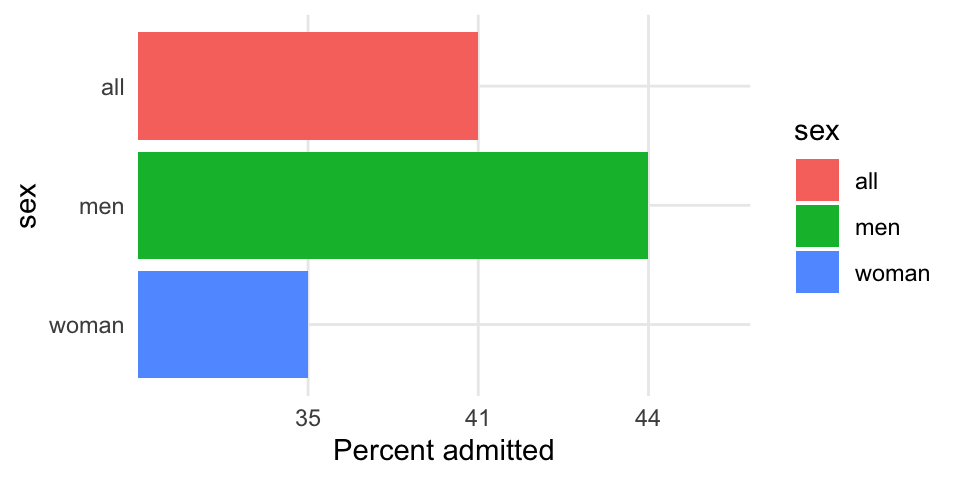
Da man dog kiggede lidt nærmere på de samme tal, men opdelte efter de forskellige afdelinger i universitet, fik man en anderledes billede af situationen. I følgende tabel har vi optagelsestallene for mænd og kvinder i hver af de forskellige afdelinger (A til F).
admissions_separate <- tribble(
~department, ~all, ~men, ~women,
#------------|-------|-------|--------#
"A", 64, 62, 82,
"B", 63, 63, 68,
"C", 35, 37, 34,
"D", 34, 33, 35,
"E", 25, 28, 24,
"F", 6, 6, 7
)Man kan man se, at for de fleste af afdelingerne, er der ikke en signifikant forskel mellem mænd og kvinder, og i nogle tilfælde havde kvinder faktisk en større chance for at blive optaget.
admissions_separate %>%
pivot_longer(-department,names_to="sex",values_to="admitted") %>%
ggplot(aes(x=department,y=admitted,fill=sex)) +
ylab("Percent admitted") +
geom_bar(stat="identity",position = "dodge",colour="black") +
theme_minimal()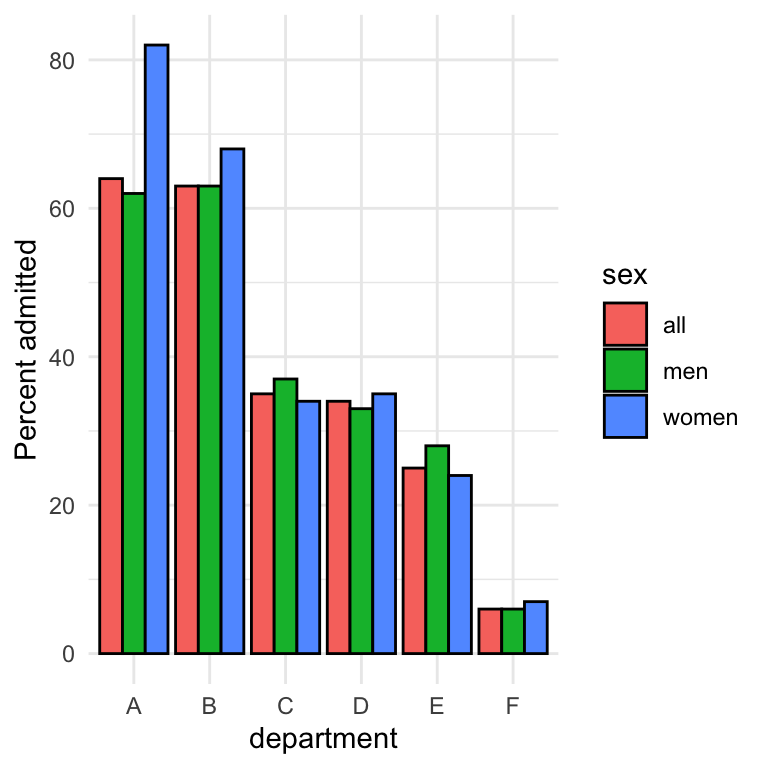
Hvad skyldes sammenhængen? Det viste sig, at kvinder havde en tendens til, at ansøge indenfor afdelingerne, som var sværeste at komme ind i. For eksempel, kan man se her, at department E har en relativt lav optagelses procent. Den samme afdeling var dog en af dem, hvor langt flere kvinder ansøgt end mænd.
applications_E <- tibble("sex"=c("all","men","woman"),applications=c("584","191","393"))
applications_E %>% ggplot(aes(x=sex,y=applications,fill=sex)) +
geom_bar(stat="identity") +
theme_minimal() +
ylab("Number of applications to dep. E") +
scale_x_discrete(limits = c("woman","men","all")) +
coord_flip()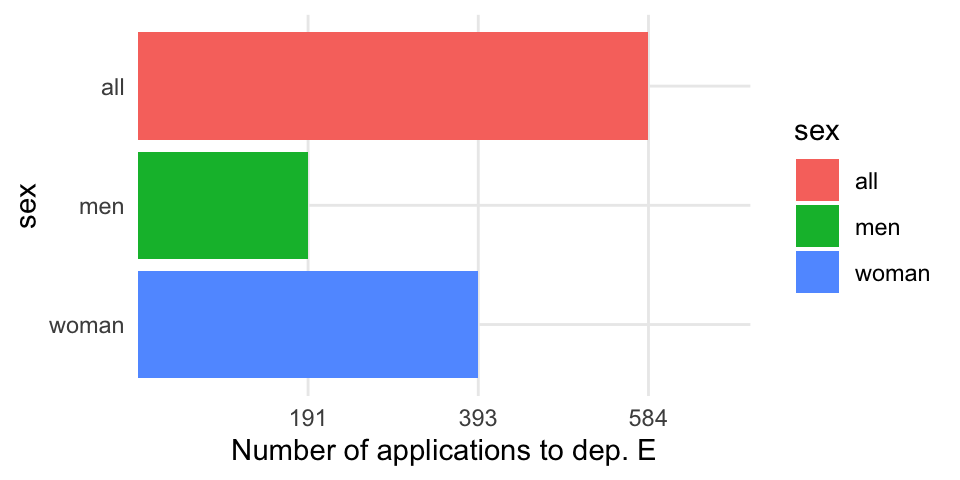
Derfor, selvom kvinder ikke havde en lavere sandsynlighed af at blive optaget end mænd i deres fortrukne fag, var antallet af kvinder der blev optaget i helhed over alle afdelinger faktisk lavere end antallet af mænd.
11.4 Case studies: Anscombe’s quartet
(Se også videoressourcer Part 3).
Anscombes quartet (se også https://en.wikipedia.org/wiki/Anscombe%27s_quartet) er et meget nyttigt og berømt eksempel, som stammer fra 1973, og som forklarer vigtigheden af at få lavet en visualisering af datasættet. Vi kan få åbnet de data fra linket nedenfor - man har x værdier og y værdier som kan anvendes til at lave et scatter plot, og der er også set, det refererer til fire forskellige datasæt (derfor ‘quartet’).
anscombe <- read.table("https://www.dropbox.com/s/mlt7crdik3eih9a/anscombe_long_format.txt?dl=1",header=T)
anscombe <- as_tibble(anscombe)
anscombe## # A tibble: 44 × 3
## set x y
## <int> <int> <dbl>
## 1 1 10 8.04
## 2 1 8 6.95
## 3 1 13 7.58
## 4 1 9 8.81
## 5 1 11 8.33
## 6 1 14 9.96
## 7 1 6 7.24
## 8 1 4 4.26
## 9 1 12 10.8
## 10 1 7 4.82
## # … with 34 more rowsFormålet med datasættet er, at man gerne vil fitte en lineær regression model for at finde den forventet y afhængig om x (husk lm(y ~ x)). Da vi har fire datasæt, kan man således opdele datasættet efter set og benytter rammen nest og map (se Chapter 7) til at fitte de fire lineær regression modeller. Vi anvender også tidy og glance for at få summary statistikker fra de fire modeller:
my_func <- ~lm(y ~ x, data = .x)
tidy_anscombe_models <- anscombe %>%
group_nest(set) %>%
mutate(fit = map(data, my_func),
tidy = map(fit, tidy),
glance = map(fit, glance))Man kan anvende unnest på den output fra tidy og kigge på den intercept og den slope af de fire modeller. Man kan se, at de to parametre er næsten identiske for de fire modeller:
tidy_anscombe_models %>% unnest("tidy") %>%
pivot_wider(id_cols = "set",names_from = "term",values_from="estimate") ## # A tibble: 4 × 3
## set `(Intercept)` x
## <int> <dbl> <dbl>
## 1 1 3.00 0.500
## 2 2 3.00 0.5
## 3 3 3.00 0.500
## 4 4 3.00 0.500Hvad med de andre parametre fra modellen - lad os kigge for eksempel på r.squared og p.value fra modellerne, som kan findes i output fra glance. Her kan vi igen se, at de er næsten identiske.
tidy_anscombe_models %>%
unnest(cols = c(glance)) %>%
select(set, r.squared,p.value)## # A tibble: 4 × 3
## set r.squared p.value
## <int> <dbl> <dbl>
## 1 1 0.667 0.00217
## 2 2 0.666 0.00218
## 3 3 0.666 0.00218
## 4 4 0.667 0.00216Hvad med korrelation? Også næsten den samme:
my_func <- ~cor(.x$x,.x$y)
anscombe %>%
group_nest(set) %>%
mutate(cor = map(data, my_func)) %>%
unnest(cor) %>%
select(-data)## # A tibble: 4 × 2
## set cor
## <int> <dbl>
## 1 1 0.816
## 2 2 0.816
## 3 3 0.816
## 4 4 0.817Kan man så konkludere, at de fire datasæt som underligger de forskellige modeller, er identiske? Vi laver et scatter plot af de fire datasæt (som vi faktisk skulle have gjort i starten af vores analyse).
anscombe %>%
ggplot(aes(x = x, y = y,colour=factor(set))) +
geom_point() +
facet_wrap(~set) +
geom_smooth(method = "lm", se = FALSE) +
theme_minimal()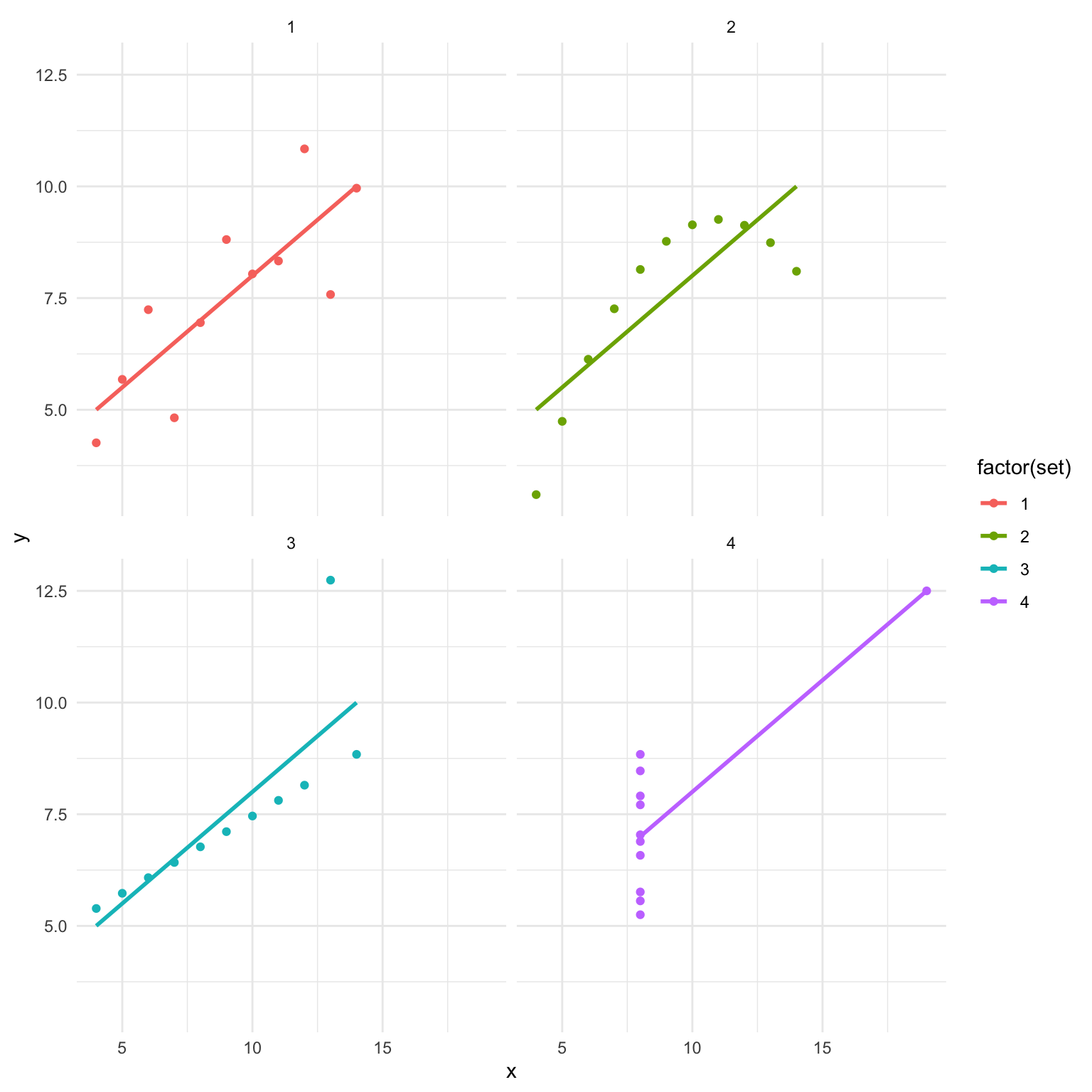
De fire datasæt er meget forskellige. Vi ved, at de har alle samme de samme beste rette linjer, men underliggende data er slet ikke de samme. Den første datasæt ser egnet til en lineær regression analyse men vi kan se i datasæt nummer to, at der ikke engang er en linæer sammenhæng. Og de andre to har outlier værdier, som gør, at den bedste rette linje passer ikke særlig god til punkterne.
11.5 Undersøgelse af “batch-effects”
(Se også videoressourcer Part 4).
Man kan også anvende visualiseringer til at kigge lidt nærmere på eventuelle batch effekter eller confounding variabler i datasættet. Det er især vigtigt i store eksperimenter, hvor forskellige samples eller dele af datasættet bliver samlede på forskellige tidspunkter, lokationer, eller af forskellige personer. Det er ofte tilfældet i sekvensering-basarede datasæt, at man ser batch effects, og det kan skyldes mange ting, bla.:
- Sekvensering dybelse
- Grupper samples lavet på forskellige tidspunkter af forskellige indivduelle
- Sekvensering maskiner - samples sekvenserne på forskellige maskiner eller ‘lanes’.
Lad os tage udgangspunkt i nogle genekspression sekvensering data lavet i mus (vi så også samme datasæt når vi lært pivot_longer kombineret med left_join).
norm.cts <- read.table("https://www.dropbox.com/s/3vhwnsnhzsy35nd/bottomly_count_table_normalised.txt?dl=1")
coldata <- read.table("https://www.dropbox.com/s/el3sm9ncvzbq6xf/bottomly_phenodata.txt?dl=1")
coldata <- as_tibble(coldata)
norm.cts <- as_tibble(norm.cts,rownames="gene")
coldata <- as_tibble(coldata)Jeg begynder ved at vælge kun rækker som har mindst 50 counts, for at undgå gener med lave ekspressionsniveauer. Det næste jeg gør er at transformere de data til log form (funktion map_if()) for at opnå en bedre fordeling i datasættet.
#normalise and filter the data
norm.cts <- norm.cts %>%
filter(rowSums(norm.cts %>% select(-gene))>50) %>%
map_if(is.numeric,~log(.x+1)) %>% as_tibble()
norm.cts## # A tibble: 10,193 × 22
## gene SRX033480 SRX033488 SRX033481 SRX033489 SRX033482 SRX033490 SRX033483
## <chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
## 1 ENSMUS… 6.35 6.32 6.21 6.29 6.31 6.27 6.30
## 2 ENSMUS… 3.51 3.56 3.57 3.27 2.99 3.61 3.56
## 3 ENSMUS… 3.19 3.50 3.08 3.21 3.14 3.09 3.22
## 4 ENSMUS… 6.69 6.48 6.38 6.35 6.39 6.34 6.50
## 5 ENSMUS… 6.05 6.37 6.17 6.26 6.16 6.06 6.13
## 6 ENSMUS… 2.89 2.94 3.16 3.21 3.77 3.30 3.11
## 7 ENSMUS… 3.42 3.12 3.86 4.36 3.77 3.99 4.24
## 8 ENSMUS… 3.42 2.94 3.52 3.41 3.57 3.60 2.99
## 9 ENSMUS… 5.02 4.98 4.49 4.27 4.67 4.35 4.81
## 10 ENSMUS… 5.13 4.88 4.97 4.76 4.82 4.79 4.96
## # … with 10,183 more rows, and 14 more variables: SRX033476 <dbl>,
## # SRX033478 <dbl>, SRX033479 <dbl>, SRX033472 <dbl>, SRX033473 <dbl>,
## # SRX033474 <dbl>, SRX033475 <dbl>, SRX033491 <dbl>, SRX033484 <dbl>,
## # SRX033492 <dbl>, SRX033485 <dbl>, SRX033493 <dbl>, SRX033486 <dbl>,
## # SRX033494 <dbl>Så der er omkring 10,000 genes i rækkerne og så 21 forskellige samples som spreder sig over kolonnerne. Vi har også nogle sample oplysninger - der er to foskellige strains af mus og også forskellige batches, som vi gerne vil kigge nærme på.
coldata## # A tibble: 21 × 5
## column num.tech.reps strain batch lane.number
## <chr> <int> <chr> <int> <int>
## 1 SRX033480 1 C57BL.6J 6 1
## 2 SRX033488 1 C57BL.6J 7 1
## 3 SRX033481 1 C57BL.6J 6 2
## 4 SRX033489 1 C57BL.6J 7 2
## 5 SRX033482 1 C57BL.6J 6 3
## 6 SRX033490 1 C57BL.6J 7 3
## 7 SRX033483 1 C57BL.6J 6 5
## 8 SRX033476 1 C57BL.6J 4 6
## 9 SRX033478 1 C57BL.6J 4 7
## 10 SRX033479 1 C57BL.6J 4 8
## # … with 11 more rowsVi kan kigger på hvor mange samples efter hver kombination af stain og batch vi har i datasættet:
table(coldata$strain, coldata$batch)##
## 4 6 7
## C57BL.6J 3 4 3
## DBA.2J 4 3 4Så kan man se, at både strain har repræsenteret tre eller fire samples i hver af de tre batches. Der er derfor replication og da vi har fået repræsenteret hver kombination af strain og batch, kan man eventuelle block efter batch for at få fjernet dens effekt. Her har vi ikke tid til at kigge på metoder for at fjerne batch-effekts, men det er vigtigt at vi er i stand til at opdage dem.
11.5.1 Principal component tilgang
Man kan undersøge mulige batch effekter via prinicpal component analysis.
pca_fit <- norm.cts %>%
select(where(is.numeric)) %>% # retain only numeric columns
prcomp(scale = TRUE) # do PCA on scaled dataHusk at når man lave en principal compononent analysis, kan man får den rotation matrix, der anvendes til at se hvor de forskellige samples ligger relative til hinanden over de forskellige principal components - dvs. at samples der ligner hinanden vises på samme sted på plottet. Den rotation matrix udtrækkes med funktionen tidy():
rot_matrix <- pca_fit %>%
tidy(matrix = "rotation") Vi vil gerne lave et plot af de rotation matrix, men første vil vi gerne tilføje de sample oplysninger med left_join, så at vi kan se de forskellige batches eller strains. Både dataframes har en kolon som hedder column, der refereres til de sample navne, så jeg forbinde efter column her.
rot_matrix <- rot_matrix %>%
left_join(coldata,by="column")Anvende pivot_wider() til at få den i wide form, så vi kan plotte “PC1” og “PC2” i et scatter plot:
rot_matrix_wide <- rot_matrix %>%
pivot_wider(names_from = "PC", names_prefix = "PC", values_from = "value")
rot_matrix_wide## # A tibble: 21 × 26
## column num.tech.reps strain batch lane.number PC1 PC2 PC3 PC4
## <chr> <int> <chr> <int> <int> <dbl> <dbl> <dbl> <dbl>
## 1 SRX03… 1 C57BL… 6 1 -0.217 0.262 0.0200 -0.508
## 2 SRX03… 1 C57BL… 7 1 -0.219 0.243 0.00586 0.166
## 3 SRX03… 1 C57BL… 6 2 -0.217 0.303 0.0153 -0.237
## 4 SRX03… 1 C57BL… 7 2 -0.219 0.246 0.00799 0.178
## 5 SRX03… 1 C57BL… 6 3 -0.217 0.259 0.0974 -0.113
## 6 SRX03… 1 C57BL… 7 3 -0.219 0.234 0.00385 0.209
## 7 SRX03… 1 C57BL… 6 5 -0.218 0.237 0.0303 -0.179
## 8 SRX03… 1 C57BL… 4 6 -0.217 0.00524 0.463 0.376
## 9 SRX03… 1 C57BL… 4 7 -0.218 0.0901 0.305 0.0651
## 10 SRX03… 1 C57BL… 4 8 -0.218 -0.110 0.434 -0.161
## # … with 11 more rows, and 17 more variables: PC5 <dbl>, PC6 <dbl>, PC7 <dbl>,
## # PC8 <dbl>, PC9 <dbl>, PC10 <dbl>, PC11 <dbl>, PC12 <dbl>, PC13 <dbl>,
## # PC14 <dbl>, PC15 <dbl>, PC16 <dbl>, PC17 <dbl>, PC18 <dbl>, PC19 <dbl>,
## # PC20 <dbl>, PC21 <dbl>Jeg giver farver og former efter de tre batches. Man kan se, at jeg har fået alle samples fra batch nummer 2 på samme sted i plottet.
rot_matrix_wide %>%
ggplot(aes(PC1,PC2,shape=factor(batch),colour=factor(batch))) +
geom_point(size=3) +
theme_minimal()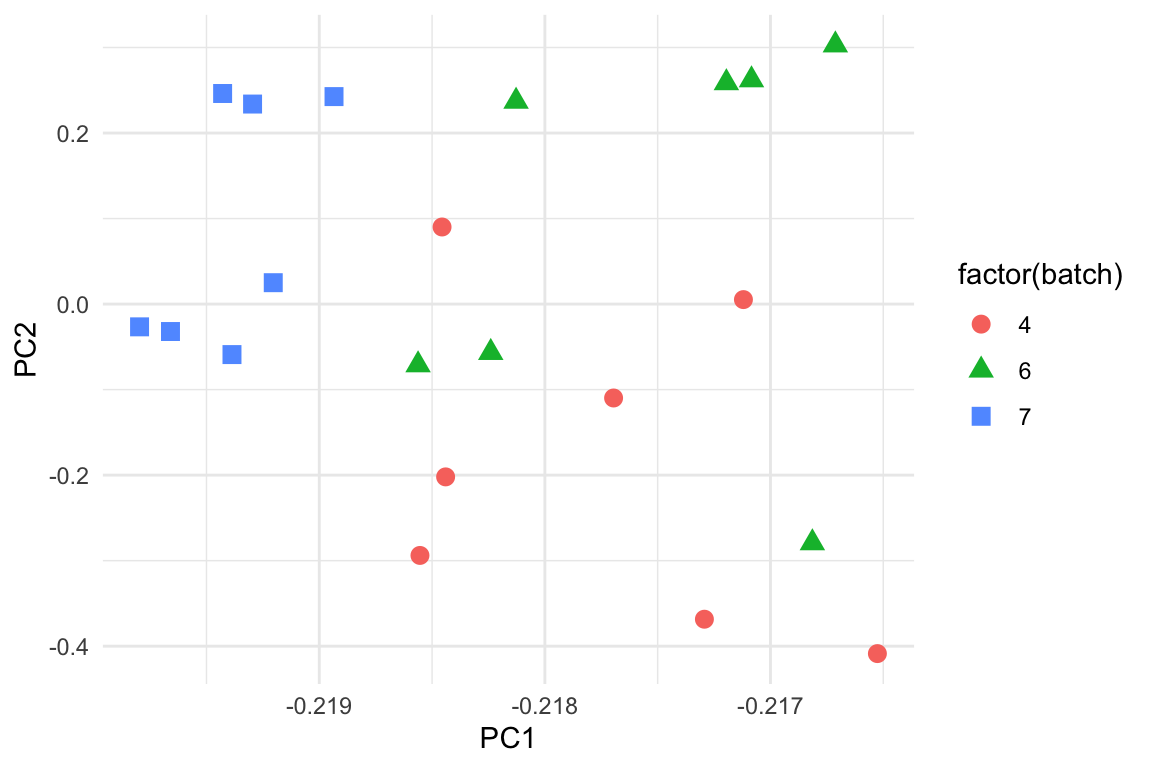
Jeg kan også give farver efter strain, hvor man kan se at der nok er en forskellen mellem de to strains her.
rot_matrix_wide %>%
ggplot(aes(PC1,PC2,shape=factor(strain),colour=factor(strain))) +
geom_point(size=3) +
theme_minimal()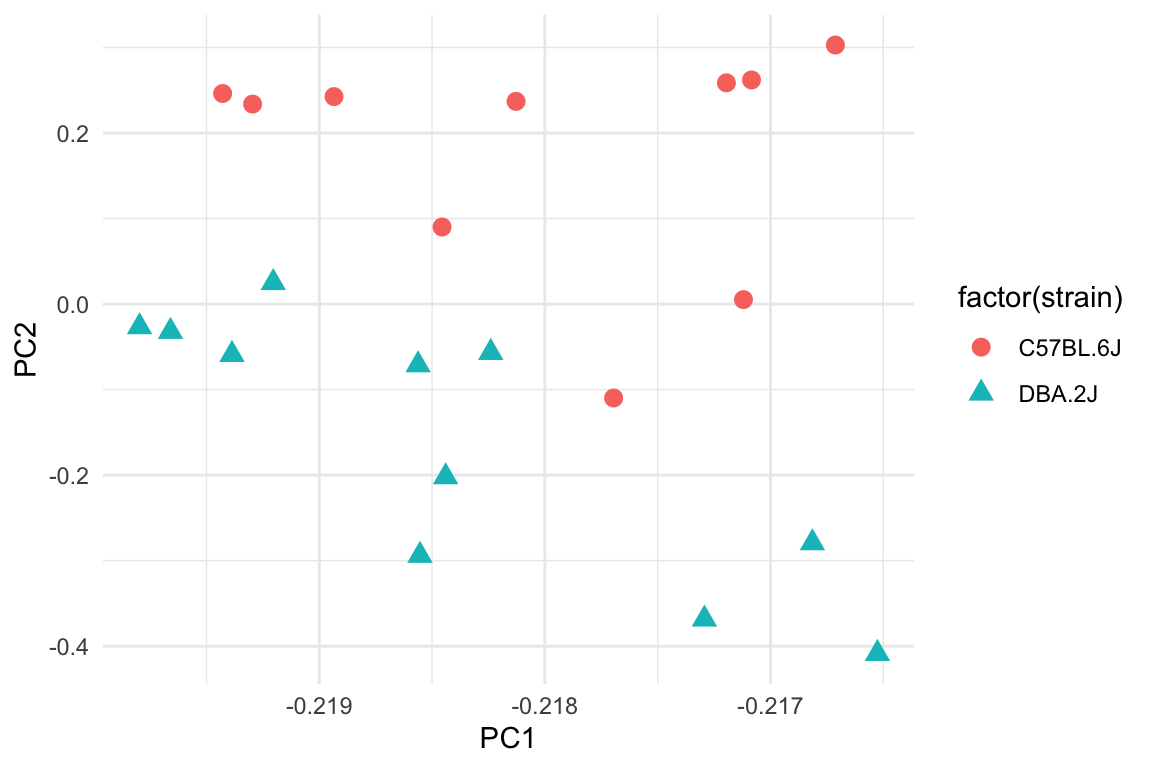
Man kan også se de data på en anden måde ved at lave boxplots for to første to principal comonponets opdelte efter batch. Vi få bekræftet vores observation at der er en stærk forskel mellem match 7 og de andre to batches langt den første principal component, og det er et problem som muligvis skal korrigeres før man laver yderligere analyser på de data.
p1 <- rot_matrix_wide %>%
ggplot(aes(x=factor(batch),y=PC1,fill=factor(batch))) + geom_boxplot(show.legend = F) + geom_jitter(show.legend = F) + theme_minimal()
p2 <- rot_matrix_wide %>%
ggplot(aes(x=factor(batch),y=PC2,fill=factor(batch))) + geom_boxplot(show.legend = F) + geom_jitter(show.legend = F) + theme_minimal()
library(gridExtra)
grid.arrange(p1,p2,ncol=2)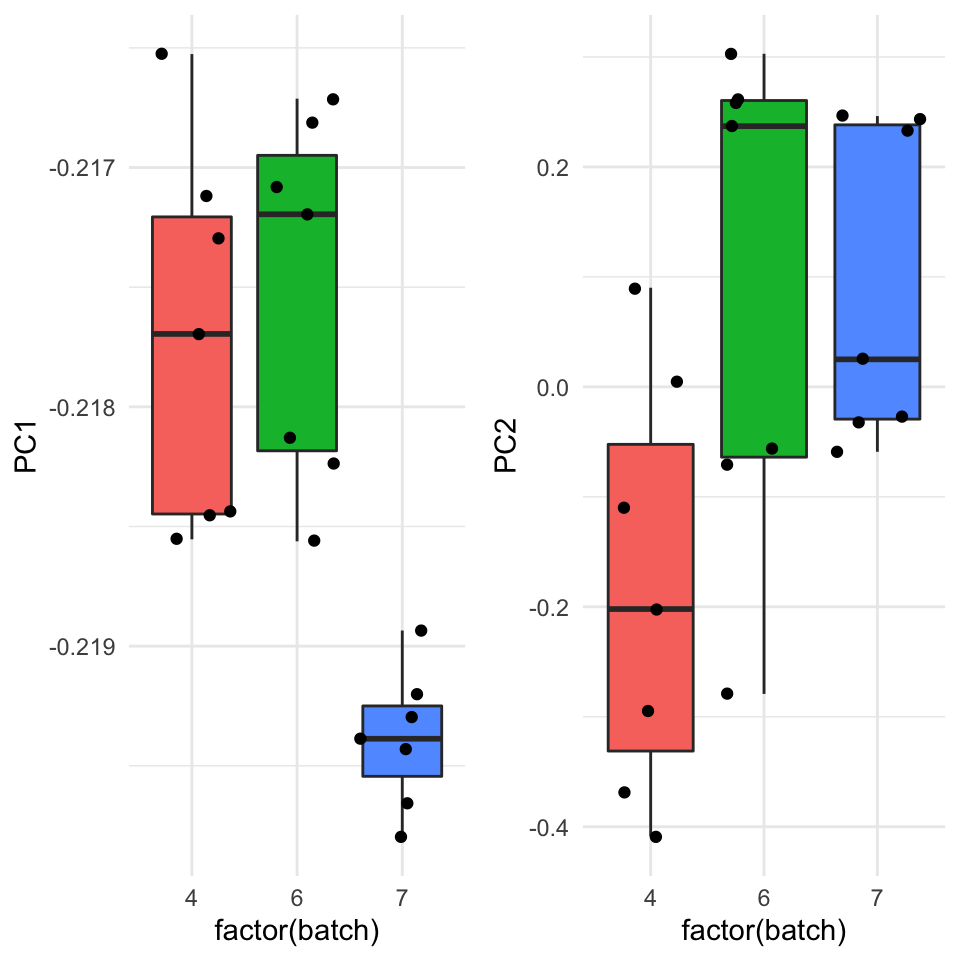
11.6 Problemstillinger
Problem 1) Quiz på Absalon - experimental
Problem 2) Eksperimental design
Jeg laver et eksperiment hvor patienter få en af tre forskellige kosttilskud (Gruppe 1, 2 og 3). Der er 5 patienter i hver gruppe og jeg vil gerne se, om patienternes energi niveau i gennemsnit er forskellige mellem de tre grupper. Alderne af patienterne i hver af de tre grupper er:
Gruppe 1: 18, 23, 31, 25, 19
Gruppe 2: 24, 29, 35, 21, 30
Gruppe 3: 43, 52, 33, 39, 40
a) Hvad er problemet her med det eksperimental design? Lav boxplots for at viser fordelingen af alderne for hver af de tre grupper.
b) Hvis man finder en signifikant forskel mellem de tre kosttilskud, kan man stoler på resultaterne?
c) Hvad andre variabler end alder kunne skyldes en eventuelle forskel mellem de tre kosttilskud, og som måske skulle tages i betragtning?
d) Hvad kan man gøre med den ovenstående eksperiment design for at løse problemet?
Problem 3) Simpson’s paradoks Lung Cap data revisited
Indlæse LungCapData og tilføj kategorisk variabel Age.Group:
LungCapData <- read.csv("https://www.dropbox.com/s/ke27fs5d37ks1hm/LungCapData.csv?dl=1")
LungCapData$Age.Group <- cut(LungCapData$Age,breaks=c(1,13,15,17,19),right=FALSE,include.lowest = TRUE)
levels(LungCapData$Age.Group) <- c("<13","13-14","15-16","17+")a) Lav boxplots med smoke på x-aksen og LungCap på y-aksen.
+ Notere hvilke gruppe har den højeste lungkapacitet.
b) Lav samme plot men adskilte efter Age.Group og beskriv, hvordan det er et eksempel på Simpson’s Paradoks.
c) Lav et boxplot med Age på y-aksen og Smoke på x-aksen for at støtte hvorfor man ser Simpson’s Paradoks i datasættet.
Problem 4) Anscombes analyse Gentage Anscombes analyse med dinosaurus datasæt:
library(datasauRus) #installer denne pakke
data_dozen <- datasauRus::datasaurus_dozen
data_dozenFALSE # A tibble: 1,846 × 3
FALSE dataset x y
FALSE <chr> <dbl> <dbl>
FALSE 1 dino 55.4 97.2
FALSE 2 dino 51.5 96.0
FALSE 3 dino 46.2 94.5
FALSE 4 dino 42.8 91.4
FALSE 5 dino 40.8 88.3
FALSE 6 dino 38.7 84.9
FALSE 7 dino 35.6 79.9
FALSE 8 dino 33.1 77.6
FALSE 9 dino 29.0 74.5
FALSE 10 dino 26.2 71.4
FALSE # … with 1,836 more rowsa) Fit en lineær regression model for hver af de datasæt (anvende group_by, nest og map ramme med en custom funktion), hvor man har y som afhængig variabel og x som uafhængig variabel. Anvend også tidy og glance på samtlige modeller
b) Anvend resultaterne fra tidy til at kigge på den slope og intercept for de forskellige modeller - ligner de hinhanden?
c) Anvende også resultaterne fra glance til at kigge på r.squared og p.value.
d) Er de alle det samme datasæt? Lav et scatter plot adskilte efter de forskellige datasæt. Hvordan ser de bedste rette linjer ud på plotterne?
Problem 5) Vi vil gerne undersøge eventuelle batch effects i følgende datasæt. Det er simuleret “single cell” sekvensering count data (dataframen cse50) samt dataframen batches, som angiver hvilken batch hver af de 500 cells tilhører.
cse50 <- read.table("https://www.dropbox.com/s/o0wzojpcsekeg6z/cell_mix_50_counts.txt?dl=1")
batches <- read.table("https://www.dropbox.com/s/4t382bfgro46ka5/cell_mix_50_batches.txt?dl=1")
batches <- as_tibble(batches)
cse50 <- as_tibble(cse50)a) Anvend map_df til at få transformeret de data til log scale (plus 1 først og tag log bagefter).
b) Lav PCA på det transformeret datasæt.
c) Anvend din PCA resultater til at få den rotation matrix
d) Forbinde oplysningerne fra dataframen batches til din rotation matrix (første tilføj en ny kolonne “column” til batches som er lig med names(cse50).
e) Anvend pivot_wider og lav et plot af den første to principal components, hvor du angiver farve efter batch.
f) Lav også boxplots for de første to prinpical components fordelt efter batch og kommenter kort på eventuelle batch effekts i de data.
Problem 6 Mere Simpson’s Paradox
Kør følgende kode til at indlæs og bearbejde følgende datasæt airlines.
airlines <- read.table("http://www.utsc.utoronto.ca/~butler/d29/airlines.txt",header=T)
airlines <- airlines %>%
pivot_longer(-airport) %>%
separate(name,sep="_",into = c("airline","status")) %>%
mutate(airline = recode(airline, aa = "Alaska", aw = "American")) %>%
pivot_wider(names_from=status,values_from=value) %>%
mutate("ontime" = ontime + delayed) %>%
rename(flights = ontime)a) Opsummer antal flights og antal delayed over de forskellige airports for at få et samlet tal til hver airline.
b) Beregn også proportionen af flights som er delayed i hver airline (igen samlede over alle airports). Lav et barplot til at vise proportionerne.
c) Denne gange opsummer over de to airlines for at få et samlet tal til hvert airport. Beregn også proportionen af flights som er delayed og lave et plot.
d) Denne gange beregne proportionen af flights som er delayed til hver kombination af både airport og airline. Igen omsæt til et plot.
e) Kan du forklare? Hint: tag for eksempel en kig på de rå data og bla. airport “Phoenix”.
11.7 Yderligere læsning
Simpson’s paradox og airlines: http://ritsokiguess.site/docs/2018/04/07/simpson-s-paradox-log-linear-modelling-and-the-tidyverse/
Batch effekt correction: https://en.wikipedia.org/wiki/Batch_effect#Correction